

Để sử dụng toàn bộ tiện ích nâng cao của Hệ Thống Pháp Luật vui lòng lựa chọn và đăng ký gói cước.
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
| Số: 03/2020/TT-BYT | Hà Nội, ngày 22 tháng 01 năm 2020 |
Căn cứ Luật số 105/2016/QH13 ngày 06 tháng 4 năm 2016 về dược;
Căn cứ Nghị định số 54/2017/NĐ-CP ngày 08 tháng 5 năm 2017 của Chính phủ quy định chi tiết một số điều và biện pháp thi hành Luật dược;
Căn cứ Nghị định số 155/2018/NĐ-CP ngày 12 tháng 11 năm 2018 của Chính phủ sửa đổi, bổ sung một số quy định liên quan đến điều kiện đầu tư kinh doanh thuộc phạm vi quản lý nhà nước của Bộ Y tế;
Căn cứ Nghị định số 75/2017/NĐ-CP ngày 20 tháng 6 năm 2017 của Chính phủ quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế;
Theo đề nghị của Cục trưởng Cục Quản lý Dược,
Bộ trưởng Bộ Y tế ban hành Thông tư sửa đổi, bổ sung một số điều của Thông tư số 11/2018/TT-BYT ngày 04 tháng 5 năm 2018 của Bộ trưởng Bộ Y tế quy định về chất lượng thuốc, nguyên liệu làm thuốc.
“Thông tư này quy định việc áp dụng tiêu chuẩn chất lượng thuốc (thuốc hóa dược, thuốc dược liệu, vắc xin, sinh phẩm), nguyên liệu làm thuốc (bao gồm cả bán thành phẩm, bán thành phẩm dược liệu, trừ dược liệu); việc kiểm nghiệm thuốc, nguyên liệu làm thuốc và thủ tục thu hồi, xử lý thuốc vi phạm.”
2. Sửa đổi, bổ sung Khoản 1 Điều 7 như sau:
“1. Áp dụng tiêu chuẩn chất lượng trong kiểm nghiệm thuốc, nguyên liệu làm thuốc:
a) Việc kiểm nghiệm phải được thực hiện theo tiêu chuẩn chất lượng thuốc, nguyên liệu làm thuốc đã được phê duyệt và cập nhật.
Trường hợp tiêu chuẩn chất lượng thuốc, nguyên liệu làm thuốc chưa được cập nhật, cơ sở kiểm nghiệm áp dụng dược điển tương ứng quy định tại Khoản 1 và Khoản 2 Điều 6 Thông tư này, tính theo ngày sản xuất lô thuốc, nguyên liệu làm thuốc được kiểm nghiệm.
Việc kiểm nghiệm thuốc pha chế, bào chế tại cơ sở khám bệnh, chữa bệnh thực hiện theo tiêu chuẩn chất lượng thuốc do cơ sở xây dựng, ban hành.
b) Trường hợp phương pháp thử nghiệm ghi trong tiêu chuẩn chất lượng thuốc, nguyên liệu làm thuốc có sai sót, không bảo đảm độ đúng, độ chính xác hoặc thử nghiệm định tính không đặc hiệu, hoặc thuốc dược liệu có nghi ngờ bổ sung thêm dược chất/chất hóa học (thuốc có phản ứng phụ, thuốc có tác dụng bất thường), hoặc thông tin về thuốc, nguyên liệu làm thuốc có chứa tạp chất từ các cơ quan quản lý dược nước ngoài, cơ sở kiểm nghiệm của Nhà nước về thuốc được áp dụng các phương pháp phân tích/kiểm nghiệm đã được quy định trong dược điển hoặc đã được thẩm định theo hướng dẫn về thẩm định phương pháp phân tích được quy định tại Phụ lục I ban hành kèm theo Thông tư số 32/2018/TT-BYT ngày 12 tháng 11 năm 2018 của Bộ trưởng Bộ Y tế quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc để kiểm nghiệm và đưa ra kết quả kiểm nghiệm chất lượng thuốc. Người đứng đầu cơ sở kiểm nghiệm thuốc phải chịu trách nhiệm về kết quả kiểm nghiệm thuốc của cơ sở mình trước pháp luật.”
3. Sửa đổi, bổ sung điểm c Khoản 3 Điều 7 như sau:
“c) Trong thời hạn tối đa 20 ngày, kể từ ngày nhận được mẫu thuốc, cơ sở kiểm nghiệm phải trả lời kết quả kiểm nghiệm, phân tích trong trường hợp sau:
- Thuốc phải kiểm nghiệm trước khi lưu hành theo quy định tại Khoản 1 Điều 8 Thông tư này, trừ trường hợp vắc xin, sinh phẩm là huyết thanh có chứa kháng thể, dẫn xuất của máu và huyết tương người theo quy định tại Khoản 2 Điều 10 Thông tư này.
- Thuốc không thuộc trường hợp quy định tại điểm b, điểm d Khoản này.”
4. Sửa đổi Khoản 6 Điều 7 như sau:
“6. Lưu hồ sơ, tài liệu:
a) Hồ sơ, tài liệu liên quan đến công tác kiểm tra chất lượng thuốc, nguyên liệu làm thuốc phải lưu giữ theo quy định tại Luật lưu trữ, Nghị định hướng dẫn Luật lưu trữ và Thông tư số 53/2017/TT-BYT ngày 29 tháng 12 năm 2017 của Bộ trưởng Bộ Y tế quy định về thời hạn bảo quản hồ sơ tài liệu chuyên môn, nghiệp vụ trong ngành y tế.
b) Hồ sơ, tài liệu khi hết thời gian lưu trữ được xử lý theo quy định của pháp luật về lưu trữ.”
5. Sửa đổi, bổ sung Khoản 3 Điều 8 như sau:
“3. Bộ Y tế (Cục Quản lý Dược) chỉ định cơ sở kiểm nghiệm đáp ứng Thực hành tốt phòng thí nghiệm (GLP) thực hiện việc kiểm nghiệm thuốc quy định tại Khoản 1 Điều này, bao gồm:
a) Cơ sở kiểm nghiệm quy định tại Khoản 1 Điều 35 của Luật Dược đáp ứng GLP, bao gồm cả cơ sở kiểm nghiệm của Nhà nước đáp ứng GLP;
b) Cơ sở dịch vụ kiểm nghiệm thuốc, nguyên liệu làm thuốc có Giấy chứng nhận đủ điều kiện kinh doanh dược với phạm vi kiểm nghiệm thuốc;
c) Cơ sở kiểm nghiệm đáp ứng GLP thuộc Cơ quan quản lý dược chặt chẽ (SRA - Stringent Regulatory Authorities) hoặc được cơ quan này chỉ định thực hiện việc kiểm nghiệm thuốc, nguyên liệu làm thuốc phục vụ cho công tác quản lý chất lượng thuốc;
d) Cơ sở kiểm nghiệm công lập cấp quốc gia được Tổ chức Y tế thế giới (WHO) đánh giá và công bố theo chương trình Tiền đánh giá Phòng kiểm nghiệm thuốc (Prequalification).
Trường hợp cơ sở kiểm nghiệm được chỉ định không đủ điều kiện để thử một hoặc một số phép thử, cơ sở kiểm nghiệm phải thông báo, niêm phong mẫu và phối hợp với cơ sở sản xuất, cơ sở nhập khẩu gửi mẫu để thử nghiệm các phép thử này tại cơ sở kiểm nghiệm khác đáp ứng GLP hoặc Phòng thử nghiệm đáp ứng tiêu chuẩn ISO/IEC 17025 và có đủ điều kiện thực hiện phép thử.”
6. Bổ sung điểm d Khoản 6 Điều 8 như sau:
“d) Lựa chọn cơ sở kiểm nghiệm đáp ứng quy định tại điểm a Khoản 3 Điều này để gửi mẫu kiểm nghiệm xác định chất lượng thuốc. Trường hợp cơ sở kiểm nghiệm không đủ điều kiện thử nghiệm một hoặc một số chỉ tiêu chất lượng, phối hợp với cơ sở kiểm nghiệm để gửi mẫu đã được cơ sở kiểm nghiệm niêm phong tới cơ sở kiểm nghiệm có đủ điều kiện thử để thử nghiệm đối với các chỉ tiêu này.”
7. Sửa đổi tiêu đề Khoản 2 Điều 10 như sau:
“Trong thời hạn tối đa là 60 ngày, kể từ ngày nhận đủ mẫu và hồ sơ theo quy định tại Điều 11 Thông tư này, Viện Kiểm định Quốc gia vắc xin và sinh phẩm y tế tiến hành:”
8. Sửa đổi, bổ sung điểm b Khoản 1 Điều 11 như sau:
“b) Mẫu vắc xin, sinh phẩm để kiểm nghiệm (số lượng mẫu đối với từng loại vắc xin, sinh phẩm theo quy định tại Hướng dẫn của Bộ Y tế về kiểm nghiệm xuất xưởng vắc xin, sinh phẩm là huyết thanh chứa kháng thể, dẫn xuất của máu và huyết tương người);”
9. Sửa đổi, bổ sung điểm b Khoản 2 Điều 11 như sau:
“b) Mẫu vắc xin, sinh phẩm để kiểm nghiệm (số lượng mẫu đối với từng loại vắc xin, sinh phẩm theo quy định tại Hướng dẫn của Bộ Y tế về kiểm nghiệm xuất xưởng vắc xin, sinh phẩm là huyết thanh chứa kháng thể, dẫn xuất của máu và huyết tương người);”
10. Bổ sung Khoản 4 Điều 11 như sau:
“4. Hồ sơ tóm tắt sản xuất và kiểm tra chất lượng của lô vắc xin, sinh phẩm được thực hiện theo hướng dẫn của WHO tại Mẫu số 09 Phụ lục III ban hành kèm theo Thông tư này.”
11. Sửa đổi, bổ sung Điều 14 như sau:
“Điều 14. Xử lý thuốc không đạt tiêu chuẩn chất lượng theo nơi lấy mẫu
1. Trường hợp mẫu thuốc vi phạm do cơ quan kiểm tra chất lượng lấy tại cơ sở bán lẻ thuốc, cơ sở khám bệnh, chữa bệnh tuyến 3, tuyến 4 (sau đây gọi chung là cơ sở bán lẻ):
a) Trong thời hạn 24 giờ, kể từ thời điểm nhận được phiếu kiểm nghiệm hoặc phiếu phân tích do cơ sở kiểm nghiệm gửi tới, Sở Y tế tiến hành niêm phong thuốc không đạt chất lượng tại cơ sở đã lấy mẫu;
b) Trong thời hạn 48 giờ, kể từ thời điểm nhận được phiếu kiểm nghiệm hoặc phiếu phân tích do cơ sở kiểm nghiệm gửi tới, Bộ Y tế (Cục Quản lý Dược) có văn bản yêu cầu cơ sở đăng ký, cơ sở sản xuất hoặc cơ sở nhập khẩu có trách nhiệm phối hợp với cơ sở phân phối bán buôn:
- Báo cáo tình hình phân phối thuốc tới cơ sở bán buôn, cơ sở khám bệnh, chữa bệnh tuyến 2 trở lên (số lượng sản xuất, nhập khẩu; tên, địa chỉ cơ sở đã mua thuốc, số lượng mua và số lượng còn tồn tại từng cơ sở) gửi về Bộ Y tế (Cục Quản lý Dược) và Sở Y tế sở tại trong thời hạn tối đa là 07 ngày kể từ ngày Bộ Y tế (Cục Quản lý Dược) ban hành văn bản yêu cầu;
- Đề nghị và phối hợp với cơ quan kiểm tra chất lượng lấy mẫu bổ sung tại cơ sở sản xuất đối với thuốc trong nước hoặc cơ sở nhập khẩu đối với thuốc nước ngoài và tại ít nhất 02 cơ sở kinh doanh sử dụng thuốc theo quy định tại Khoản 4 Điều này; gửi báo cáo kết quả thực hiện về Bộ Y tế (Cục Quản lý Dược) trong thời hạn tối đa là 15 ngày kể từ ngày Bộ Y tế (Cục Quản lý Dược) ban hành văn bản yêu cầu;
- Gửi mẫu đã lấy tới cơ sở kiểm nghiệm tuyến Trung ương để kiểm tra chất lượng đối với chỉ tiêu không đạt.
c) Căn cứ kết quả kiểm nghiệm các mẫu thuốc được lấy bổ sung, Cục Quản lý Dược xử lý theo quy định tại Khoản 5 Điều này.
2. Trường hợp mẫu thuốc vi phạm do cơ quan kiểm tra chất lượng lấy tại cơ sở bán buôn, cơ sở khám bệnh, chữa bệnh tuyến 2 trở lên (sau đây gọi là cơ sở bán buôn):
a) Trong thời hạn 24 giờ, kể từ thời điểm nhận được phiếu kiểm nghiệm hoặc phiếu phân tích do cơ sở kiểm nghiệm gửi tới, Sở Y tế tiến hành niêm phong thuốc không đạt chất lượng tại cơ sở đã lấy mẫu;
b) Trong thời hạn 48 giờ, kể từ thời điểm nhận được phiếu kiểm nghiệm hoặc phiếu phân tích do cơ sở kiểm nghiệm gửi tới, Bộ Y tế (Cục Quản lý Dược) xác định mức độ vi phạm và kết luận về việc thu hồi thuốc vi phạm theo quy định tại Phụ lục II ban hành kèm theo Thông tư này và ban hành văn bản:
- Thông báo thu hồi thuốc trên địa bàn tỉnh, thành phố trực thuộc Trung ương nơi lấy mẫu và cơ sở kinh doanh, sử dụng đã được cơ sở bán buôn nơi lấy mẫu thuốc cung cấp theo quy định tại Khoản 3, Khoản 4 Điều 12 Thông tư này;
- Yêu cầu cơ sở đăng ký, cơ sở sản xuất hoặc cơ sở nhập khẩu có trách nhiệm phối hợp với cơ sở phân phối bán buôn:
+ Báo cáo tình hình phân phối thuốc tới cơ sở bán buôn (số lượng sản xuất, nhập khẩu; tên, địa chỉ cơ sở đã mua thuốc, số lượng mua và số lượng còn tồn tại từng cơ sở) gửi về Bộ Y tế (Cục Quản lý Dược) và Sở Y tế sở tại trong thời hạn tối đa là 07 ngày kể từ ngày Bộ Y tế (Cục Quản lý Dược) ban hành văn bản;
+ Đề nghị và phối hợp với cơ quan kiểm tra chất lượng lấy mẫu bổ sung ít nhất 02 mẫu thuốc tại cơ sở kinh doanh sử dụng thuốc theo quy định tại Khoản 4 Điều này; gửi báo cáo kết quả thực hiện về Bộ Y tế (Cục Quản lý Dược) trong thời hạn tối đa là 15 ngày kể từ ngày Bộ Y tế (Cục Quản lý Dược) ban hành văn bản yêu cầu;
+ Gửi mẫu đã lấy tới cơ sở kiểm nghiệm tuyến Trung ương để kiểm tra chất lượng đối với chỉ tiêu không đạt.
c) Căn cứ kết quả kiểm nghiệm các mẫu thuốc được lấy bổ sung, Cục Quản lý Dược xử lý theo quy định tại Khoản 5 Điều này.
3. Trường hợp mẫu thuốc do cơ quan kiểm tra chất lượng lấy tại cơ sở sản xuất, cơ sở nhập khẩu, cơ sở kinh doanh dịch vụ bảo quản thuốc hoặc mẫu thuốc được xác định vi phạm chất lượng do nguyên nhân trong quá trình sản xuất, hoặc trường hợp lô thuốc đã được lấy mẫu đồng thời tại 02 cơ sở bán buôn, Bộ Y tế (Cục Quản lý Dược) xác định mức độ vi phạm và kết luận về việc thu hồi thuốc vi phạm theo quy định tại Phụ lục II ban hành kèm theo Thông tư này, ban hành quyết định thu hồi thuốc theo quy định tại Khoản 3 Điều 12 Thông tư này. Phạm vi và thời gian thu hồi thực hiện theo quy định tại Khoản 3 Điều 63 Luật Dược.
4. Yêu cầu đối với việc lấy mẫu bổ sung để kiểm tra chất lượng theo quy định tại Khoản 1 và Khoản 2 Điều này:
Cơ quan kiểm tra chất lượng thuốc xác định phương án lấy mẫu trên cơ sở báo cáo về tình hình phân phối của cơ sở sản xuất, cơ sở nhập khẩu; ưu tiên lấy mẫu theo thứ tự a, b, c, d, đ được quy định sau đây:
a) Các mẫu thuốc được lấy tại các cơ sở bán buôn tại các địa bàn tỉnh, thành phố khác nhau; trong đó có cơ sở bán buôn đã cung cấp thuốc cho cơ sở đã được lấy mẫu;
b) Các mẫu thuốc được lấy tại các cơ sở bán buôn tại các địa bàn tỉnh, thành phố khác nhau;
c) Các mẫu thuốc được lấy tại các cơ sở bán buôn tại cùng một địa bàn tỉnh, thành phố;
d) Các mẫu thuốc được lấy tại cơ sở bán buôn và tại cơ sở bán lẻ.
đ) Các mẫu thuốc được lấy tại cơ sở bán lẻ.
e) Chỉ áp dụng lấy mẫu theo phương án đ khi cơ sở sản xuất, cơ sở nhập khẩu chứng minh thuốc không còn được bảo quản, tồn trữ tại cơ sở bán buôn. Không lấy mẫu bổ sung đối với số thuốc đã thu hồi.
5. Xử lý kết quả kiểm nghiệm mẫu thuốc được lấy bổ sung.
a) Trường hợp các mẫu thuốc được lấy bổ sung đạt tiêu chuẩn chất lượng, Bộ Y tế (Cục Quản lý Dược) có văn bản xác định mức độ vi phạm, cơ sở chịu trách nhiệm về vi phạm và chỉ đạo Sở Y tế xử lý đối với thuốc của cơ sở bán lẻ đã lấy mẫu ban đầu đối với trường hợp quy định tại Khoản 1 Điều này hoặc cơ sở bán buôn và thuốc đã thu hồi trên địa bàn tỉnh, thành phố đối với trường hợp quy định tại Khoản 2 Điều này.
Phạm vi và thời gian thu hồi thực hiện theo quy định tại Khoản 3 Điều 63 Luật dược;
b) Trường hợp ít nhất 01 mẫu thuốc được lấy bổ sung tại cơ sở bán lẻ không đạt tiêu chuẩn chất lượng, trừ trường hợp quy định tại điểm a Khoản này, Bộ Y tế (Cục Quản lý Dược) đánh giá nguy cơ, có văn bản xác định mức độ vi phạm, cơ sở chịu trách nhiệm về vi phạm, chỉ đạo Sở Y tế xử lý đối với thuốc tại các cơ sở bán lẻ đã lấy mẫu và cảnh báo về điều kiện bảo quản và chất lượng của thuốc.”
c) Trường hợp ít nhất 01 (một) mẫu thuốc được lấy bổ sung tại cơ sở bán buôn hoặc tất cả các mẫu thuốc được lấy bổ sung tại cơ sở bán lẻ theo quy định tại điểm đ Khoản 4 Điều này không đạt tiêu chuẩn chất lượng, Bộ Y tế (Cục Quản lý Dược) xác định mức độ vi phạm và kết luận về việc thu hồi thuốc vi phạm theo quy định tại Phụ lục II ban hành kèm theo Thông tư này, ban hành quyết định thu hồi thuốc theo quy định tại Khoản 3 Điều 12 Thông tư này.”
12. Bổ sung điểm d Khoản 2 Điều 15 như sau:
“d) Thuốc giả, thuốc nhập lậu, thuốc không rõ nguồn gốc, xuất xứ, thuốc hết hạn dùng, thuốc có chứa các chất bị cấm sử dụng, thuốc sản xuất từ nguyên liệu không đạt tiêu chuẩn chất lượng, thuốc thuộc trường hợp phải bị tiêu hủy theo quy định tại Nghị định về xử phạt hành chính trong lĩnh vực y tế, mẫu thuốc lưu đã hết thời gian lưu theo quy định.
13. Sửa đổi, bổ sung Khoản 6 Điều 15 như sau:
“6. Hủy thuốc:
a) Người đứng đầu cơ sở có thuốc bị tiêu hủy ra quyết định thành lập Hội đồng hủy thuốc để tổ chức việc hủy thuốc, quyết định phương pháp hủy, giám sát việc hủy thuốc. Hội đồng có ít nhất 03 người, trong đó phải có 01 đại diện là người phụ trách chuyên môn của cơ sở;
b) Việc hủy thuốc phải đảm bảo an toàn cho người, súc vật và tránh ô nhiễm môi trường theo các quy định của pháp luật về bảo vệ môi trường.
Cơ sở có thuốc bị tiêu hủy phải chịu toàn bộ trách nhiệm liên quan đến việc hủy thuốc và phải báo cáo kèm theo biên bản hủy thuốc tới Sở Y tế sở tại theo quy định tại mẫu số 06 Phụ lục III ban hành kèm theo Thông tư này.
c) Quy định về việc hủy vắc xin:
- Tối thiểu 07 ngày trước khi thực hiện việc tiêu hủy vắc xin, cơ sở hủy vắc xin phải có văn bản thông báo kế hoạch hủy đến Sở Y tế sở tại, trong đó phải có các thông tin về tên, số lượng, nồng độ hoặc hàm lượng của từng vắc xin cần hủy, lý do xin hủy, thời gian hủy, địa điểm hủy và phương pháp hủy. Sở Y tế có trách nhiệm giám sát việc hủy vắc xin.
- Quy trình hủy vắc xin và việc hủy vắc xin phải được thực hiện theo đúng các quy định hiện hành tại Thông tư liên tịch số 58/2015/TTLT-BYT-BTNMT ngày 31/12/2015 của Bộ trưởng Bộ Y tế và Bộ trưởng Bộ Tài nguyên và Môi trường quy định về quản lý chất thải y tế và Thông tư số 36/2015/TT-BTNMT ngày 30/6/2015 của Bộ trưởng Bộ Tài nguyên và Môi trường về quản lý chất thải nguy hại.- Trong thời hạn 07 ngày kể từ ngày kết thúc việc hủy vắc xin, cơ sở phải có văn bản báo cáo việc hủy vắc xin kèm theo biên bản hủy tới Sở Y tế sở tại và Cục Quản lý Dược. Biên bản hủy theo quy định tại mẫu số 06 Phụ lục III ban hành kèm theo Thông tư này.
d) Việc hủy thuốc phải kiểm soát đặc biệt phải thực hiện theo quy định tại Điều 48 của Nghị định số 54/2017/NĐ-CP.”
14. Sửa đổi, bổ sung điểm b và điểm e Khoản 1 Điều 18 như sau:
“b) Chủ trì phối hợp với Viện Kiểm nghiệm thuốc Trung ương, Viện Kiểm nghiệm thuốc thành phố Hồ Chí Minh, Viện Kiểm định Quốc gia vắc xin và sinh phẩm y tế xây dựng kế hoạch lấy mẫu thuốc để kiểm tra chất lượng tại các cơ sở sản xuất, pha chế, nhập khẩu, xuất khẩu, bảo quản, bán buôn, bán lẻ và sử dụng trên phạm vi cả nước, trình Bộ Y tế xem xét, phê duyệt và bố trí ngân sách thực hiện kế hoạch theo thẩm quyền.
Triển khai việc lấy mẫu thuốc để kiểm tra chất lượng theo kế hoạch đã được phê duyệt và cập nhật vào hệ thống dữ liệu thông tin kiểm tra chất lượng thuốc của Bộ Y tế các thông tin về mẫu thuốc, nguyên liệu làm thuốc được lấy (bao gồm các thông tin: tên thuốc, nguyên liệu làm thuốc, nồng độ, hàm lượng, dạng bào chế, số lô, hạn dùng, số giấy đăng ký lưu hành hoặc giấy phép nhập khẩu, cơ sở sản xuất, cơ sở nhập khẩu, cơ sở lấy mẫu) và kết quả kiểm tra chất lượng đối với mẫu thuốc, nguyên liệu làm thuốc;”
“e) Chủ trì, phối hợp với các cơ quan chức năng liên quan chịu trách nhiệm dịch, công bố và cập nhật trên Trang thông tin điện tử của Cục Quản lý Dược tài liệu hướng dẫn của Tổ chức Y tế thế giới (WHO) về hủy thuốc để các cơ sở tham khảo trong quá trình lựa chọn phương pháp hủy và thực hiện hủy.”
15. Sửa đổi, bổ sung điểm b Khoản 2 Điều 18 như sau:
“b) Xây dựng kế hoạch lấy mẫu thuốc, nguyên liệu làm thuốc để kiểm tra chất lượng tại cơ sở sản xuất, pha chế, nhập khẩu, xuất khẩu, bảo quản, bán buôn, bán lẻ và sử dụng trên địa bàn tỉnh, thành phố, trình Ủy ban nhân dân tỉnh, thành phố trực thuộc Trung ương xem xét, phê duyệt và bố trí ngân sách thực hiện kế hoạch theo thẩm quyền;”.
16. Sửa đổi, bổ sung điểm a Khoản 3 Điều 18 như sau:
“a) Cơ sở kiểm nghiệm thuốc tuyến Trung ương (Viện Kiểm nghiệm thuốc Trung ương, Viện Kiểm nghiệm thuốc thành phố Hồ Chí Minh, Viện Kiểm định Quốc gia vắc xin và sinh phẩm y tế):
- Thực hiện phân tích, kiểm nghiệm mẫu để xác định chất lượng thuốc, nguyên liệu làm thuốc sản xuất, lưu hành, sử dụng; báo cáo kết quả kiểm nghiệm về Bộ Y tế (Cục Quản lý Dược) và Sở Y tế nơi lấy mẫu;
- Nghiên cứu, thiết lập và công bố trên trang thông tin điện tử của các Viện và của Cục Quản lý Dược danh mục các chất chuẩn, chất đối chiếu, tạp chất chuẩn phục vụ cho việc phân tích, kiểm nghiệm mẫu thuốc, nguyên liệu làm thuốc được sản xuất, nhập khẩu, lưu hành, sử dụng trên lãnh thổ Việt Nam;
- Viện Kiểm nghiệm thuốc Trung ương, Viện Kiểm nghiệm thuốc thành phố Hồ Chí Minh chịu trách nhiệm cung cấp cho Trung tâm kiểm nghiệm thuốc tỉnh, thành phố trực thuộc Trung ương, theo địa bàn được phân công, bản sao hoặc văn bản điện tử của tiêu chuẩn chất lượng thuốc, nguyên liệu làm thuốc;
- Viện Kiểm định Quốc gia vắc xin và sinh phẩm y tế, theo định kỳ hàng năm, rà soát, đánh giá xu hướng chất lượng vắc xin, sinh phẩm, gửi Cục Quản lý Dược rà soát trình Bộ Y tế ban hành Hướng dẫn của Bộ Y tế về kiểm nghiệm xuất xưởng vắc xin, sinh phẩm là huyết thanh chứa kháng thể, dẫn xuất của máu và huyết tương người; nội dung bao gồm:
+ Chính sách chung về kiểm nghiệm xuất xưởng, bao gồm cả các chính sách miễn giảm thử nghiệm đối với vắc xin, sinh phẩm đã được Cơ quan quản lý dược nghiêm ngặt (SRA -Stringent Regulatory Authorities) đánh giá, cấp chứng nhận xuất xưởng lô (Batch Release Certificate).
+ Chỉ tiêu phải thử nghiệm khi kiểm nghiệm để cấp giấy chứng nhận chất lượng, thời gian cấp giấy chứng nhận chất lượng đối với từng sản phẩm vắc xin, sinh phẩm; và
+ Mẫu Hồ sơ tóm tắt sản xuất và kiểm tra chất lượng của lô vắc xin, sinh phẩm cho từng loại vắc xin, sinh phẩm.
Cập nhật thông tin về việc cấp giấy chứng nhận chất lượng vắc xin, sinh phẩm là huyết thanh chứa kháng thể, dẫn xuất của máu và huyết tương của người trên trang thông tin điện tử của Viện và Cục Quản lý Dược.”
17. Bổ sung điểm a Khoản 5 Điều 18 như sau:
Thu hồi phí lấy mẫu do cơ sở kinh doanh hoàn trả và chi phí kiểm nghiệm đối với mẫu thuốc, nguyên liệu làm thuốc không đạt tiêu chuẩn chất lượng theo quy định của pháp luật”.
18. Bổ sung điểm b Khoản 5 Điều 18 như sau:
“- Thu hồi phí lấy mẫu do cơ sở kinh doanh hoàn trả và chi phí kiểm nghiệm đối với mẫu thuốc, nguyên liệu làm thuốc không đạt tiêu chuẩn chất lượng theo quy định của pháp luật.”
19. Bổ sung sửa đổi khoản 8 Mục I Phụ lục I như sau:
“8. Lấy mẫu dược liệu
1. Dược liệu hoặc dược liệu đã được chế biến một phần, kể cả động vật, thực vật (cây thuốc đã làm khô và các phần của cây) và khoáng chất, được coi như nguyên liệu không đồng đều, lấy mẫu theo quy định tại Mục I, khoản 9, sơ đồ r của Phụ lục này.
2. Lấy mẫu để giám sát chất lượng dược liệu của cơ quan kiểm tra chất lượng thuốc nhà nước: Thực hiện theo quy định tại “Hướng dẫn kiểm tra chất lượng dược liệu” năm 2011 của Tổ chức Y tế thế giới (Quality control methods for herbal materials 2011). Trường hợp lô dược liệu không đồng nhất, việc lấy mẫu thực hiện theo quy định điểm 1 khoản này.”
20. Bổ sung Mẫu số 09 Phụ lục III Hồ sơ tóm tắt sản xuất và kiểm tra chất lượng của lô vắc xin, sinh phẩm kèm theo Thông tư này.
Thông tư này có hiệu lực từ ngày 16 tháng 3 năm 2020
Điều 3. Trách nhiệm thi hành.
Cục trưởng Cục Quản lý Dược, Chánh Văn phòng Bộ, Chánh Thanh tra Bộ Thủ trưởng các đơn vị thuộc và trực thuộc Bộ Y tế, Sở Y tế các tỉnh, thành phố trực thuộc Trung ương, các cơ sở kinh doanh dược và các cơ quan, tổ chức, cá nhân khác có liên quan chịu trách nhiệm thi hành Thông tư này.
Trong quá trình thực hiện nếu có khó khăn, vướng mắc đề nghị các cơ quan tổ chức, cá nhân phản ánh về Bộ Y tế (Cục Quản lý Dược) để xem xét, giải quyết./.
|
| KT. BỘ TRƯỞNG |
HƯỚNG DẪN VIỆC LẤY MẪU THUỐC, NGUYÊN LIỆU LÀM THUỐC ĐỂ XÁC ĐỊNH CHẤT LƯỢNG
(Kèm theo Thông tư số 11/2018/TT-BYT ngày 04 tháng 5 năm 2018 của Bộ trưởng Bộ Y tế)
I. Trình tự lấy mẫu và các thao tác lấy mẫu
1. Dụng cụ lấy mẫu thuốc, nguyên liệu làm thuốc
Dụng cụ lấy mẫu, đồ đựng mẫu phải được làm bằng vật liệu trơ, sạch thích hợp với đặc điểm của từng loại mẫu, đảm bảo không làm ảnh hưởng đến chất lượng mẫu, không đưa tạp chất vào mẫu gây ô nhiễm, nhiễm chéo đối với mẫu cũng như phải đảm bảo an toàn cho người lấy mẫu (Tham khảo Mục III).
2. Lượng mẫu cần lấy
2.1. Lượng mẫu cần lấy để phân tích và để lưu được tính toán tùy thuộc vào yêu cầu kiểm tra, tiêu chuẩn chất lượng thuốc, nguyên liệu làm thuốc áp dụng, phương pháp thử của mẫu nhưng ít nhất phải đủ cho ba lần phân tích hoặc phải đủ để thực hiện các phép thử đảm bảo thu được kết quả chính xác và tin cậy.
2.2. Thông thường, mỗi lô sản xuất được lấy hai mẫu (một mẫu phân tích và một mẫu lưu tại cơ quan kiểm nghiệm). Trường hợp cần thiết, số mẫu phân tích và mẫu lưu có thể nhiều hơn hai để đủ gửi kiểm nghiệm và lưu ở các cơ quan, tổ chức có liên quan.
3. Thao tác lấy mẫu
3.1. Nguyên tắc lấy mẫu:
- Tùy theo mục đích kiểm tra và theo từng loại sản phẩm, người lấy mẫu quyết định lựa chọn phương pháp lấy mẫu thích hợp.
- Quá trình lấy mẫu phải được giám sát và được ghi chép lại đầy đủ. Tất cả các dấu hiệu không đồng nhất, hư hỏng của thuốc và bao bì bảo quản đều phải được ghi chép lại.
- Quy trình lấy mẫu phải đảm bảo sao cho có thể kịp thời phát hiện tính không đồng nhất của thuốc trong từng đơn vị lấy mẫu và của cả lô thuốc. Các dấu hiệu không đồng nhất bao gồm sự khác nhau về hình dạng, kích thước, hoặc màu sắc của các tiểu phân chất rắn ở dạng kết tinh, dạng hạt hoặc dạng bột; lớp vỏ ấm của các chất hút có tính hút ẩm; sự lắng đọng các dược chất ở dạng rắn trong thuốc dạng chất lỏng hoặc bán rắn; sự tách lớp của thuốc dạng chất lỏng.
- Không trộn lẫn, phối hợp các mẫu được lấy từ các phần có dấu hiệu khác nhau, từ các bao bì có nghi ngờ chất lượng của lô thuốc, vì sự trộn lẫn này làm che khuất các dấu hiệu tạp nhiễm, hàm lượng thấp hoặc các vấn đề chất lượng khác. Phải tạo thành mẫu riêng biệt từ các phần, các bao bì này.
- Đối với thành phẩm thuốc, quy trình lấy mẫu cần tính đến các phép thử chính thức và phép thử bổ sung đối với từng dạng thuốc (ví dụ: thuốc viên nén, hoặc thuốc tiêm truyền...). Các phép thử bổ sung bao gồm các phép thử để xác định thuốc giả mạo, thuốc bị pha trộn, thuốc thêm các chất không được phép.
- Không nên trộn lại thuốc đã lấy ra khỏi bao bì trực tiếp với thuốc còn trong bao bì.
3.2. Trình tự lấy mẫu
- Kiểm tra tình trạng vật lý của lô hàng: phân tách theo từng loại sản phẩm và từng lô sản xuất, mỗi lô lại tách riêng các thùng hàng có dấu hiệu bị hư hại, không đảm bảo vệ sinh để kiểm tra, lấy mẫu riêng. Loại bỏ các đơn vị bao gói không có nhãn.
- Từ lô sản phẩm lấy ra các đơn vị lấy mẫu, mở các bao gói để lấy các mẫu ban đầu và làm kín ngay lại các bao gói đã được lấy mẫu. Số lượng nguyên liệu trong mẫu ban đầu được tính toán đủ để chuẩn bị mẫu tiếp sau.
- Trộn đều các mẫu ban đầu thành những mẫu riêng của từng đơn vị lấy mẫu.
- Trộn đều các mẫu riêng thành một mẫu chung.
- Tạo mẫu cuối cùng: Từ mẫu chung lấy ra các phần bằng nhau tạo thành mẫu cuối cùng gồm mẫu phân tích và mẫu lưu.
3.3. Các mẫu phân tích và mẫu lưu phải được cho vào đồ đựng, hàn kín và dán nhãn. Nhãn của đồ đựng mẫu phải ghi rõ tên thuốc, tên nhà sản xuất, ký hiệu lô sản xuất, hạn dùng, số thùng đã lấy mẫu, nơi lấy mẫu, số lượng mẫu đã lấy (nếu mẫu lấy là nguyên liệu thuốc gây nghiện, hướng thần, tiền chất dùng làm thuốc và nguyên liệu thuốc phóng xạ số lượng cần phải ghi bằng chữ), ngày lấy mẫu, các điều kiện bảo quản phù hợp với biên bản lấy mẫu.
3.4. Sau khi lấy mẫu xong, các thành viên tham gia lấy mẫu phải niêm phong riêng biệt mẫu phân tích và mẫu lưu để đảm bảo mẫu được an toàn trong quá trình vận chuyển từ nơi lấy mẫu đến nơi giao mẫu. Trên niêm phong của mẫu phải ghi rõ ngày tháng lấy mẫu và có ít nhất chữ ký của người lấy mẫu và đại diện cơ sở được lấy mẫu.
Trong trường hợp cần thiết, phần còn lại sau khi lấy mẫu cũng phải niêm phong để đề phòng sự tráo mẫu thuốc, nguyên liệu làm thuốc.
3.5. Lập biên bản lấy mẫu: biên bản lấy mẫu phải ghi rõ số lô, ngày lấy mẫu, địa điểm lấy mẫu, các điều kiện bảo quản, ghi chép về bất cứ nhận xét nào khác liên quan và những bất thường của quá trình lấy mẫu, có ít nhất tên và chữ ký của người lấy mẫu và đại diện cơ sở được lấy mẫu.
Trong trường hợp đoàn kiểm tra chất lượng tiến hành lấy mẫu thì phải có thêm chữ ký của Trưởng đoàn kiểm tra.
Trong trường hợp đại diện cơ sở được lấy mẫu không ký biên bản, thì biên bản có chữ ký của người lấy mẫu và người chứng kiến.
Biên bản phải làm thành ít nhất ba bản: một bản lưu tại cơ sở được lấy mẫu, một bản lưu ở cơ quan kiểm nghiệm, một bản lưu tại cơ quan quản lý, kiểm tra chất lượng thuốc.
4. Lấy mẫu nguyên liệu làm thuốc
4.1. Trường hợp nguyên liệu chỉ có một bao gói:
a) Lấy mẫu nguyên liệu dạng rắn: Lấy mẫu ban đầu ở các vị trí khác nhau của thùng hàng (phía trên, giữa và đáy). Nếu các mẫu ban đầu không có các dấu hiệu cảm quan khác nhau thì trộn đều các mẫu ban đầu thành mẫu riêng.
b) Lấy mẫu nguyên liệu dạng lỏng hoặc bán rắn: Nếu không đồng đều thì phải trộn đều trước khi lấy mẫu. Ví dụ nếu chế phẩm lỏng phân lớp phải khuấy đều trước khi lấy mẫu, hoặc nếu có cặn lắng trong chất lỏng phải làm tan cặn lắng hoặc phân tán đều trước khi lấy mẫu bằng cách có thể làm ấm hoặc khuấy trộn đều.
4.2. Trường hợp lô nguyên liệu có nhiều bao gói:
Tùy theo mục đích của lấy mẫu kiểm tra, mức độ đồng nhất và chất lượng của lô thuốc mà chọn phương án lấy mẫu thích hợp theo quy định tại Mục I, khoản 9 của Phụ lục này.
5. Lấy mẫu bán thành phẩm chưa đóng gói
Các sản phẩm loại này là thuốc bột, thuốc nước, siro thuốc, thuốc mỡ, thuốc cốm, thuốc viên, thuốc tiêm... chứa trong các bao gói lớn để chuyển đến cơ sở đóng gói lẻ. Mỗi lô sản xuất được lấy mẫu theo cách sau:
1. Nếu lô sản phẩm chỉ có 1 - 2 bao gói, thì mở cả hai bao gói. Nếu lô sản phẩm có từ 3 bao gói trở lên thì mở ba bao gói. Lấy ít nhất 3 mẫu ban đầu ở các vị trí khác nhau của mỗi bao gói.
2. Trộn các mẫu ban đầu lại thành mẫu chung rồi tạo mẫu cuối cùng gồm mẫu phân tích và mẫu lưu.
6. Lấy mẫu vật liệu bao gói
Lấy mẫu vật liệu bao gói thực hiện theo quy định tại Mục I, khoản 9 của Phụ lục này.
7. Lấy mẫu thuốc thành phẩm
7.1. Lấy mẫu thuốc thành phẩm để kiểm tra hoặc giám sát chất lượng:
a) Việc lấy mẫu theo nguyên tắc lấy mẫu ngẫu nhiên và phải lấy mẫu ở những vị trí khác nhau của lô hàng.
b) Căn cứ tiêu chuẩn chất lượng thuốc, số lượng thuốc được lấy sao cho đủ để thử nghiệm và lưu mẫu. Trường hợp không có đủ thông tin để tính toán chính xác số lượng thuốc cần lấy, tham khảo số lượng thuốc thành phẩm tối thiểu cần lấy theo quy định tại Mục V của Phụ lục này.
c) Trình tự lấy mẫu được thực hiện trên cơ sở hướng dẫn tại Mục II của Phụ lục này.
7.2. Lấy mẫu để kiểm tra cảm quan khi nhập thuốc: số lượng mẫu lấy để kiểm tra cảm quan theo quy định tại Mục IV của Phụ lục này.
8. Lấy mẫu dược liệu
1. Dược liệu hoặc dược liệu đã được chế biến một phần, kể cả động vật, thực vật (cây thuốc đã làm khô và các phần của cây) và khoáng chất, được coi như nguyên liệu không đồng đều, lấy mẫu theo quy định tại Mục I khoản 9, sơ đồ r của Phụ lục này.
2. Lấy mẫu để giám sát chất lượng dược liệu của cơ quan kiểm tra chất lượng thuốc nhà nước: Thực hiện theo quy định tại “Hướng dẫn kiểm tra chất lượng dược liệu” năm 2011 của Tổ chức Y tế thế giới (Quality control methods for herbal materials 2011). Trường hợp lô dược liệu không đồng nhất, việc lấy mẫu thực hiện theo quy định điểm 1 khoản này.
9. Sơ đồ lấy mẫu nguyên liệu ban đầu và vật liệu bao gói
9.1. Trước khi thực hiện việc lấy mẫu, người lấy mẫu phải kiểm tra tính nguyên vẹn, mức độ hư hỏng của thùng đựng, sự đồng đều của sản phẩm bên trong của mỗi đơn vị lấy mẫu.
9.2. Việc lấy mẫu có thể được thực hiện theo một trong ba sơ đồ lấy mẫu ghi tại Bảng 1 dưới đây.
Bảng 1: Các giá trị n, p hoặc r cho N đơn vị bao gói
| Giá trị n, p, r | Giá trị N | ||
| Sơ đồ n | Sơ đồ p | Sơ đồ r | |
| 2 | Tới 3 | Tới 25 | Tới 2 |
| 3 | 4 - 6 | 25 - 56 | 3 - 4 |
| 4 | 7 - 13 | 57 - 100 | 5 - 7 |
| 5 | 14 - 20 | 101 - 156 | 8 - 11 |
| 6 | 21 - 30 | 157 - 225 | 12 - 16 |
| 7 | 31 - 42 |
| 17 - 22 |
| 8 | 43 - 56 |
| 23 - 28 |
| 9 | 57 - 72 |
| 29 - 36 |
| 10 | 73 - 90 |
| 37 - 44 |
a) Sơ đồ n
Sử dụng “Sơ đồ n” trong trường hợp lô nguyên liệu cần lấy mẫu được coi là đồng nhất và được cung cấp từ một nguồn xác định. Có thể lấy mẫu từ bất kỳ phần nào trong thùng nguyên liệu (thường từ lớp trên cùng). “Sơ đồ n” dựa trên công thức n = 1 +![]() , với N là số đơn vị bao gói của lô hàng, số đơn vị lấy mẫu tối thiểu n có được bằng cách làm tròn đơn giản. Từ n đơn vị lấy mẫu được chọn ngẫu nhiên, lấy ra các mẫu ban đầu, đựng trong các đồ đựng mẫu riêng biệt. Nếu các mẫu ban đầu lấy được không có nghi ngờ gì về cảm quan và định tính, các mẫu ban đầu được trộn đều thành mẫu riêng, mẫu chung để chia thành mẫu phân tích và mẫu lưu theo trình tự chung.
, với N là số đơn vị bao gói của lô hàng, số đơn vị lấy mẫu tối thiểu n có được bằng cách làm tròn đơn giản. Từ n đơn vị lấy mẫu được chọn ngẫu nhiên, lấy ra các mẫu ban đầu, đựng trong các đồ đựng mẫu riêng biệt. Nếu các mẫu ban đầu lấy được không có nghi ngờ gì về cảm quan và định tính, các mẫu ban đầu được trộn đều thành mẫu riêng, mẫu chung để chia thành mẫu phân tích và mẫu lưu theo trình tự chung.
b) Sơ đồ p
Sử dụng “sơ đồ p” trong trường hợp lô nguyên liệu được xem là đồng nhất, từ một nguồn xác định và mục đích chính là để kiểm tra định tính. “Sơ đồ p” dựa vào công thức p = 0,4![]() , với N là số đơn vị bao gói của lô hàng. Giá trị p có được bằng cách làm tròn lên đến số nguyên lớn nhất tiếp theo. Các mẫu ban đầu được lấy từ mỗi trong số N đơn vị bao gói của lô hàng và được đựng trong các đồ đựng mẫu riêng biệt. Các mẫu ban đầu này được kiểm tra về cảm quan, định tính. Nếu kết quả phù hợp, p mẫu chung được tạo thành bằng cách trộn lẫn thích hợp các mẫu ban đầu để lưu hoặc phân tích (nếu cần thiết).
, với N là số đơn vị bao gói của lô hàng. Giá trị p có được bằng cách làm tròn lên đến số nguyên lớn nhất tiếp theo. Các mẫu ban đầu được lấy từ mỗi trong số N đơn vị bao gói của lô hàng và được đựng trong các đồ đựng mẫu riêng biệt. Các mẫu ban đầu này được kiểm tra về cảm quan, định tính. Nếu kết quả phù hợp, p mẫu chung được tạo thành bằng cách trộn lẫn thích hợp các mẫu ban đầu để lưu hoặc phân tích (nếu cần thiết).
c) Sơ đồ r
Sử dụng “sơ đồ r” khi lô nguyên liệu bị nghi ngờ là không đồng nhất và/hoặc tiếp nhận từ nguồn không xác định, dược liệu hay các nguyên liệu ban đầu là dược liệu đã được chế biến một phần. Sơ đồ này dựa trên công thức r = 1,5![]() , với N là số đơn vị bao gói của lô sản phẩm. Giá trị r thu được bằng cách làm tròn tới số nguyên lớn nhất tiếp theo.
, với N là số đơn vị bao gói của lô sản phẩm. Giá trị r thu được bằng cách làm tròn tới số nguyên lớn nhất tiếp theo.
Các mẫu ban đầu được lấy từ mỗi trong số N đơn vị bao gói và được đựng trong các đồ đựng mẫu riêng biệt. Các mẫu ban đầu này được kiểm tra cảm quan và định tính. Nếu kết quả phù hợp, lựa chọn ngẫu nhiên r mẫu để thực hiện kiểm nghiệm riêng rẽ. Nếu kết quả kiểm nghiệm đồng nhất, các mẫu lưu có thể được gộp lại thành 01 mẫu lưu.
9.3. Lấy mẫu nguyên liệu ban đầu để định tính đối với các cơ sở sản xuất không áp dụng các sơ đồ trên mà theo nguyên tắc “Thực hành tốt sản xuất thuốc” theo khuyến cáo của Tổ chức Y tế thế giới (GMP-WHO).
II. Các bước thực hiện lấy mẫu
1. Các chế phẩm lỏng chờ đóng gói
Các bước cần được xem xét khi lấy mẫu các chế phẩm lỏng chờ đóng gói như sau:
- Đọc và hiểu các khuyến cáo thận trọng để đảm bảo an toàn khi cấp phát nguyên liệu.
- Tập trung các thiết bị lấy mẫu cần thiết (ống lấy mẫu hay bình lấy mẫu có thể cân được weighted sampling, các bình đựng mẫu lấy và nhãn) và kiểm tra để đảm bảo tất cả những dụng cụ cần thiết đều sạch.
- Xác định vị trí của lô.
- Kiểm tra các đồ đựng xem có dấu hiệu ô nhiễm lô hay không. Ghi lại bất cứ điểm nghi ngờ nào.
- Kiểm tra các nhãn để phát hiện những khác biệt rõ ràng và những dấu hiệu thay đổi, kể cả tẩy xóa và ghi nhãn nhầm. Ghi lại bất cứ điểm nghi ngờ nào.
- Tìm hiểu và làm rõ các nguồn gốc gây sai sót vì bất kỳ lý do gì trước khi tiến hành.
- Chọn ống lấy mẫu chế phẩm lỏng có cỡ và miệng phù hợp với độ nhớt của chế phẩm lỏng cần lấy mẫu.
- Lấy mẫu chế phẩm lỏng, hỗn dịch hay nhũ tương (đã được khuấy đều, nếu thích hợp) bằng cách ấn từ từ ống lấy mẫu để mở vào chế phẩm lỏng theo phương thẳng đứng sao cho lấy được sản phẩm từ mỗi lớp.
- Đóng chặt ống mẫu, rút ống mẫu ra khỏi chế phẩm lỏng và để cho chế phẩm dính bên ngoài ống được róc hết. Chuyển toàn bộ mẫu đã lấy trong ống sang một bình đựng mẫu sạch và có dán nhãn.
- Lặp lại các bước trên cho đến khi lấy đủ mẫu để phân tích và để lưu.
- Niêm phong bình đựng mẫu.
- Niêm phong lại thùng sản phẩm vừa lấy mẫu và dán nhãn “đã lấy mẫu”.
- Làm sạch và khô ống lấy mẫu, lưu ý những thận trọng về an toàn.
- Tiếp tục lấy mẫu ở những thùng sản phẩm khác theo cách tương tự các bước ở trên.
- Làm sạch ống lấy mẫu bằng quy trình làm sạch đã quy định.
- Chuyển các mẫu phân tích tới phòng kiểm nghiệm và các mẫu lưu đến kho lưu mẫu. Báo cáo lại bất cứ điểm nghi ngờ nào liên quan tới việc lấy mẫu mà người phân tích và thanh tra viên cần lưu ý.
- Kiểm tra giấy chứng nhận phân tích của nhà cung cấp so với các tiêu chuẩn, nếu có.
2. Nguyên liệu ban đầu dạng bột
Các bước cần thực hiện khi lấy mẫu nguyên liệu ban đầu dạng bột như sau:
- Đọc và hiểu các lưu ý thận trọng cần thực hiện để đảm bảo an toàn khi xử lý nguyên liệu.
- Tập hợp các thiết bị lấy mẫu (xiên lấy mẫu, bình đựng mẫu lấy và nhãn) và kiểm tra xem tất cả có sạch hay không.
- Xác định đợt hàng và đếm số thùng
- Kiểm tra tất cả các thùng xem có gì khác và có dấu hiệu bị hư hỏng không. Ghi lại bất cứ điểm nghi ngờ nào.
- Kiểm tra tất cả các nhãn xem có khác hay có dấu hiệu thay đổi nào không, kể cả tẩy xóa và ghi nhầm nhãn. Ghi lại bất cứ điểm nghi ngờ nào.
- Tách các thùng bị hư hỏng và những thùng mà sản phẩm bên trong nghi bị hỏng để kiểm tra riêng. Sau đó những thùng này phải được đề cập hay bị từ chối.
- Tách riêng các thùng có số lô khác và xử lý riêng.
- Đánh số những thùng còn lại.
- Chọn sơ đồ lấy mẫu thích hợp (n, p hoặc r).
- Chọn những thùng cần lấy mẫu theo yêu cầu của sơ đồ đã được chọn (dùng bảng số ngẫu nhiên, vẽ sơ đồ lô hay dùng đánh số ngẫu nhiên).
- Mỗi lần mở một thùng và kiểm tra sản phẩm bên trong. Ghi lại nếu có khác biệt.
- Chọn xiên lấy mẫu thích hợp và sạch, xiên (với cửa lấy mẫu đóng kín) vào bột thuốc sao cho đầu xiên chạm đáy thùng.
- Mở cửa để lấy bột thuốc vào các khoang xiên, sau đó đóng lại
- Rút xiên ra khỏi thùng đựng mẫu và chuyển xiên đã chứa bột thuốc được lấy sang một bình đựng mẫu đã dán nhãn
- Lặp lại các bước trên cho đến khi lấy đủ vật liệu để phân tích và lưu.
- Niêm phong bình đựng mẫu lấy.
- Niêm phong lại thùng sản phẩm vừa lấy mẫu và dán nhãn có ghi “đã lấy mẫuˮ.
- Lau sạch xiên lấy mẫu nếu cần, chú ý những thận trọng về an toàn, trước khi lấy mẫu các thùng khác.
- Lặp lại các bước như trên với mỗi thùng đã chọn.
- Lau sạch xiên lấy mẫu dùng quy trình làm sạch đã quy định.
- Chuyển các mẫu phân tích tới phòng kiểm nghiệm và các mẫu lưu đến kho lưu mẫu. Báo cáo lại bất cứ nghi ngờ nào liên quan tới lấy mẫu mà người phân tích và thanh tra viên cần lưu ý.
- Kiểm tra giấy chứng nhận phân tích của nhà cung cấp so với các tiêu chuẩn, nếu có.
3. Nguyên liệu bao gói
Các bước cần xem xét thực hiện khi lấy mẫu nguyên liệu đóng gói như sau:
- Kiểm tra đợt hàng so với hồ sơ liên quan.
- Kiểm tra các thùng trung chuyển theo các chi tiết sau và báo cáo bất cứ sự chênh lệch nào nếu cần:
+ Xác định các thông tin đúng;
+ Niêm phong còn nguyên vẹn, nếu có niêm phong;
+ Không bị hư hỏng.
- Lấy mẫu cần thiết từ số thùng nguyên liệu theo yêu cầu, đặc biệt lưu ý đến những quy định về lấy mẫu nguyên liệu đóng gói ở Mục I, khoản 9 của Phụ lục này.
- Đưa mẫu đã lấy vào các đồ đựng mẫu thích hợp.
- Đánh dấu các thùng nguyên liệu đã được lấy mẫu.
- Lưu ý bất kỳ tình huống đặc biệt nào xảy ra trong quá trình lấy mẫu (ví dụ: hàng kém chất lượng hay các thành phần bị hư hại). Báo cáo những bất thường quan sát được.
- Chuyển tất cả các pa-lét hay thùng nguyên liệu đã lấy mẫu khỏi khu vực lấy mẫu cùng với toàn bộ hồ sơ.
- Kiểm tra giấy chứng nhận phân tích của nhà cung cấp so với các tiêu chuẩn, nếu có.
4. Thuốc thành phẩm
Khi lấy mẫu thành phẩm cần cân nhắc các bước sau:
- Xác định số pa-lét cho mỗi lô trong đợt hàng.
- Tính toán số pa-lét dựa theo số đơn vị lấy mẫu để kiểm tra mẫu bằng cảm quan quy định:
+ Kiểm tra điều kiện của pa-lét và bao bì về tính toàn vẹn của nguyên liệu đóng gói bên ngoài.
+ Kiểm tra phía ngoài của hàng hóa trên các pa-lét xem có sạch không.
+ Kiểm tra xem ghi nhãn trên các pa-lét có phù hợp với danh mục hàng hóa đóng gói không.
+ Đếm, phân loại và ghi chép các sai sót.
- Tính tổng số gói (hộp) đựng hàng vận chuyển trên số pa-lét hiện có và xác minh tổng số căn cứ vào danh mục đóng gói hàng.
- Kiểm tra các gói (hộp) hàng vận chuyển (đơn vị đóng gói trung gian) từ số pa-lét đã chọn:
+ Kiểm tra xem nguyên liệu đóng gói của các hộp đựng hàng có còn nguyên vẹn không.
+ Kiểm tra xem các hộp có sạch không.
+ Kiểm tra xem nhãn trên các hộp có bị hư hỏng không.
+ Kiểm tra các hộp xem có hư hỏng gì không.
+ Kiểm tra các nhãn xem có lỗi chính tả không.
+ Kiểm tra ngày sản xuất và hạn dùng trên các nhãn.
+ Đếm, phân loại và ghi chép các sai sót.
- Kiểm tra bằng cảm quan các đơn vị đóng gói cuối cùng từ các đơn vị đóng gói trung gian:
+ Kiểm tra xem vật liệu đóng gói của các đơn vị đóng gói cuối cùng có còn nguyên vẹn không.
+ Kiểm tra xem các gói hàng có sạch không.
+ Kiểm tra hình dạng và màu sắc của các gói hàng.
+ Kiểm tra xem nhãn trên các gói hàng có bị hư hỏng không.
+ Kiểm tra các gói xem có hư hỏng gì không.
+ Kiểm tra các nhãn xem có lỗi chính tả không.
+ Kiểm tra ngày sản xuất và hạn dùng trên các nhãn.
+ Đếm, phân loại và ghi chép số lỗi.
- Từ số gói hàng được chọn, dựa theo tiêu chuẩn chất lượng, bằng phương pháp lấy mẫu ngẫu nhiên, tính số gói hàng cần kiểm tra lý học, hóa học và số lượng cần cho mẫu lưu.
- Kiểm tra giấy xác nhận phân tích của nhà cung cấp so với các tiêu chuẩn, nếu có.

Hình 1. Các loại xẻng lấy mẫu chế phẩm rắn

Hình 2. Tuýp lấy mẫu chế phẩm lỏng và chế phẩm bôi ngoài da
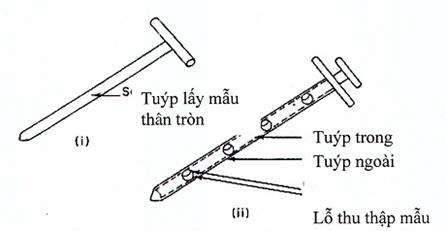
Hình 3. Lấy mẫu từ các đồ đựng nguyên liệu răn sâu lỏng
(i) Tuýp lấy mẫu thân tròn (Hình 3.i): gồm một ống rỗng với một thanh bên trong có một đầu nhọn để chọc được vào bột ở một vị trí kín. Đầu của thanh bên trong thường nhọn để ít ảnh hưởng tới lớp bột. Một số loại có khóa cho phép ước lượng mẫu lấy ở mức cần thiết, nên sẽ giảm được chênh lệch về khối lượng giữa các mẫu lấy.
(ii) Tuýp lấy mẫu kép: (Hình 3.ii): gồm hai ống đồng tâm, ống bên trong làm bằng vật liệu cứng trừ các khoang đựng mà mẫu được lấy vào đó, ống bên ngoài rỗng có lỗ hổng có thể khớp với các khoang đựng mẫu ở ống bên trong, đầu nhọn để giảm thiểu tình trạng làm vỡ đối với lớp bột.
Chú ý: Khi đưa dụng cụ này vào một hỗn hợp bột tĩnh sẽ làm xáo trộn hỗn hợp bột, làm di chuyển bột thuốc từ các lớp trên xuống các lớp dưới. Mức độ xáo trộn tùy thuộc vào động tác đưa dụng cụ theo kiểu đưa từ từ hay giật cục hay xoáy. Do đó cán bộ phải được đào tạo sử dụng kỹ thuật thích hợp.
Góc đưa dụng cụ vào khối bột thuốc cũng có thể ảnh hưởng tới mẫu lấy được. Nếu đưa dụng cụ lấy mẫu vào khối bột thuốc theo phương thẳng đứng thì có thể lấy được mẫu với kích thước tiểu phân khác so với những mẫu lấy được khi dùng cùng dụng cụ đó nhưng được đưa vào khối bột theo một góc nhọn. Ngoài ra, mẫu lấy được cũng bị ảnh hưởng bởi vị trí của buồng đựng mẫu của dụng cụ lấy mẫu so với khối bột thuốc (tức là buồng đựng mẫu ở vị trí đỉnh, đáy hoặc ở giữa của dụng cụ lấy mẫu).
Mẫu lấy được cũng bị ảnh hưởng bởi độ dày (độ sâu) của bao bì đựng thuốc, do bột nguyên liệu thuốc bị đẩy vào các buồng đựng mẫu bởi lực nén tĩnh của khối bột thuốc. Lực nén ở dưới đáy của thùng lớn lớn hơn nhiều so với lực nén ở lớp giữa hay ở trên cùng. Do vậy, với cùng một dụng cụ lấy mẫu, có khả năng lấy được các phần của mẫu có kích thước tiểu phân bột thuốc khác nhau ở lớp trên và lớp đáy của khối bột thuốc tĩnh.
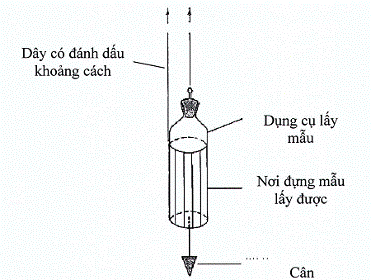
Hình 4. Dụng cụ lấy mẫu có thể cân được (Weighted container)
Lấy mẫu từ các bể chứa và thùng chứa lớn thì dùng loại dụng cụ lấy mẫu có thể cân được. Đồ đựng này được thiết kế sao cho có thể mở ra ở một độ sâu cần thiết. Các điểm đánh dấu trên dây được dùng để xác định khi nào thì dụng cụ đựng tới độ sâu phù hợp.
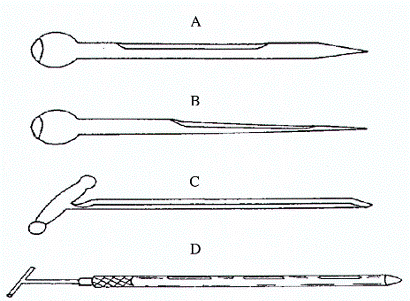
Hình 5. Các loại xiên lấy mẫu đơn giản
A: Xiên lấy mẫu đóng, được sử dụng lấy mẫu có kích thước hạt lớn như sắn
B: Xiên lấy mẫu đóng, được sử dụng lấy mẫu có kích thước hạt nhỏ
C: Xiên lấy mẫu mở
D: Xiên lấy mẫu hai tuýp
Các loại xiên để lấy mẫu từ các túi sản phẩm, để dễ dàng đưa dụng cụ lấy mẫu vào túi sản phẩm xiên lấy mẫu thường có hình vót thon, đường kính ngoài khoảng 12 mm, nhưng có thể tới 25 mm, dài khoảng 40 - 45 cm.
| Cỡ lô Số đơn vị bao gói thương phẩm/lô | Số đơn vị bao gói thương phẩm cần lấy cho một mẫu kiểm tra |
| Từ 2 đến 8 | 2 |
| 9 - 15 | 3 |
| 16 - 25 | 5 |
| 26 - 50 | 8 |
| 51 - 90 | 13 |
| 91 - 150 | 20 |
| 151 - 280 | 32 |
| 281 - 500 | 50 |
| 501 - 1200 | 80 |
| 1201 - 3200 | 125 |
| 3201 - 10000 | 200 |
| 10001 - 35000 | 315 |
| 35001 - 150000 | 500 |
| 150001 - 500000 | 600 |
| 500001 trở lên | 1250 |
V. Cơ số mẫu lấy để kiểm tra chất lượng
Số mẫu thuốc, nguyên liệu làm thuốc được lấy để kiểm tra chất lượng (chưa bao gồm mẫu để lưu) được quy định như sau:
| STT | Dạng bào chế | Chủng loại, quy cách | Số lượng |
| 1 | Thuốc viên nén, viên nang, viên bao | 1 hoạt chất | 80 viên |
| ≥ 2 hoạt chất | 120 viên | ||
| 2 | Thuốc nước | ≥ 100 ml | 20 chai (lọ) |
| 10 - 100 ml | 30 chai (lọ) | ||
| 5ml - 10ml | 50 chai (lọ) | ||
| < 5ml | 100 chai (lọ) | ||
| 3 | Cốm, bột | Đóng gói theo đơn vị đơn liều hoặc đa liều | ~100 gam |
| Hoàn cứng, hoàn mềm | > 0,5 g/viên | 120 viên | |
| 0,1 - 0,5 g/viên | 200 viên | ||
| < 0,1 g/viên | 500 viên | ||
| 4 | Rượu thuốc | ≤ 650 ml | 7 chai |
| > 650 ml | 5 chai | ||
| 5 | Dịch truyền | ≥ 250 ml | 20 chai |
| 100 ml - 250 ml | 25 chai | ||
| < 100 ml | 50 chai | ||
| Ống tiêm | 1ml | 150 ống | |
| ≥ 2 ml | 120 ống | ||
| Nước cất tiêm | 2 ml | 250 ống | |
| 5 ml | 100 ống | ||
| 10 ml | 80 ống | ||
| 6 | Thuốc nhỏ mắt | ≤ 2ml/100mg | 100 lọ (tuýp) |
| > 2ml/100mg | 80 lọ (tuýp) | ||
| 7 | Thuốc mỡ, kem, gel dùng ngoài | ≤ 100mg | 30 lọ (tuýp) |
| > 100mg | 40 lọ (tuýp) | ||
| 8 | Thuốc bột tiêm | < 100 mg | 150 lọ |
| 100 - 450 mg | 120 lọ | ||
| > 450 mg | 100 lọ | ||
| 9 | Dầu xoa | 1 - 2 ml | 30 lọ |
| ≥ 5 ml | 20 lọ | ||
| 10 | Cao thuốc | Các loại | ~100g |
| 11 | Dược liệu | Chứa tinh dầu | 250 g |
| Không chứa tinh dầu | 100 g | ||
| 12 | Tinh dầu | Các loại | 150 ml |
| 13 | Vắc xin, sinh phẩm | Các loại | Theo quy định của nhà sản xuất |
| 14 | Nguyên liệu | Nguyên liệu quý | 20 g |
| Nguyên liệu kháng sinh | 50 g | ||
| Nguyên liệu thuốc gây nghiện, hướng thần, tiền chất | 10g | ||
| Nguyên liệu thường | 100 g | ||
| Nhựa hạt | 200 g | ||
| 15 | Dây truyền dịch | Các loại | 30 bộ |
| 16 | Ống thủy tinh rỗng | 2 ml | 500 ống |
| ≥ 5 ml | 300 ống | ||
| 17 | Chai đựng dịch truyền | Các loại | 10 chai |
BIỂU MẪU
(Kèm theo Thông tư số 11/2018/TT-BYT ngày 04 tháng 5 năm 2018 của Bộ trưởng Bộ Y tế)
Mẫu số 09: Mẫu hồ sơ tóm tắt quá trình sản xuất và kiểm tra chất lượng của lô vắc xin
Mỗi hồ sơ tóm tắt lô được gắn với một sản phẩm cụ thể, nhưng bắt buộc phải bao gồm các nội dung thông tin chung như sau:
| Đầu mục | Thông tin cơ bản | Tiêu chí cần rà soát |
| Định danh của nhà sản xuất | Tên nhà sản xuất | Đảm bảo khả năng truy nguyên và nhận dạng |
| Số đăng ký | Số đăng ký duy nhất | Đảm bảo khả năng truy nguyên và nhận dạng |
| Địa điểm sản xuất | Địa điểm sản xuất từng bán thành phẩm, bán thành phẩm cuối cùng và thành phẩm | Đảm bảo khả năng truy nguyên và nhận dạng |
| Tên và số lô | Tên và số lô thành phẩm, bán thành phẩm, bán thành phẩm cuối cùng và dung môi pha loãng nếu có. | Tính duy nhất, tính hệ thống, đảm bảo khả năng truy nguyên và nhận dạng |
| Cỡ lô | Dung tích, số liều và loại đơn vị chứa (VD: lọ) | Các thông tin liệt kê phải phù hợp với giới hạn cho phép. |
| Hạn dùng | Với mỗi nguyên liệu đầu (nếu có), bán thành phẩm trung gian, bán thành phẩm cuối cùng và thành phẩm | Hạn dùng của từng thành phần. |
| Ngày sản xuất | Với mỗi nguyên liệu đầu quan trọng (VD: lô chủng, ngân hàng tế bào, nguyên liệu có nguồn gốc động vật ...), bán thành phẩm trung gian, bán thành phẩm cuối cùng và thành phẩm. | So sánh với hạn dùng đã ghi để tính toán và xác nhận. |
| Lưu đồ | Lưu đồ quy trình sản xuất bao gồm các thông tin truy nguyên của các thành phần chính, kể cả số lô. | Xác nhận về nhận dạng và tính logic của luồng nguyên liệu, bán thành phẩm trung gian, bán thành phẩm cuối cùng và thành phẩm. |
| Chủng giống và tế bào | Tên, số lô, số đời cấy truyền. | Chủng giống cho sản xuất và loại tế bào, số đời cấy truyền của chủng/tế bào gốc và/hoặc chủng/tế bào làm việc giống như hồ sơ đăng ký đã được NRA phê duyệt hoặc được khuyến cáo bởi WHO. |
| Quy trình sản xuất | Mỗi giai đoạn sản xuất (VD: nuôi cấy, tinh chế, bất hoạt), quy trình thử nghiệm cũng như tiêu chuẩn và kết quả thu được; số lô của các bán thành phẩm trung gian và số lượng/dung tích, điều kiện bảo quản. | Đảm bảo chúng đáp ứng tiêu chuẩn đã được duyệt. Hiệu suất của các giai đoạn quan trọng nằm trong giới hạn cho phép. |
| Công thức bào chế | Số lượng các thành phần hoạt tính trong công thức bào chế, kèm theo số lô, và dung tích các bán thành phẩm, điều kiện bảo quản. | Xác nhận lại số liệu thực tế và việc tính toán dựa trên các thông tin đã cung cấp. |
| Các thử nghiệm | Kết quả thực tế của các thử nghiệm trên nguyên liệu, bán thành phẩm trung gian, bán thành phẩm cuối cùng và thành phẩm, kèm theo tiêu chuẩn của chúng; bao gồm từng thử nghiệm riêng lẻ và kết quả trung bình; Cung cấp ngày bắt đầu thử nghiệm, phương pháp tiến hành, danh sách các mẫu chuẩn, tham chiếu, hóa chất thuốc thử quan trọng và tình trạng hiệu lực của chúng, kèm theo đánh giá hiệu năng của các mẫu chuẩn, tham chiếu và mẫu chứng, VD: các tiêu chí đánh giá tính phù hợp của phép thử hàm lượng (assay) (độ dốc, điểm gốc, tính tuyến tính, 50% endpoint, kết quả nội chuẩn, liều thử thách); Cung cấp các kết quả thống kê như trung bình, trung bình nhân, độ lệch chuẩn, khoảng tin cậy 95%... nếu có; kể cả các kết quả thử nghiệm không đạt, không đúng và phải thực hiện lại. | Chứng minh rằng các đặc tính nhận dạng, tinh sạch, an toàn, hiệu lực và ổn định nhiệt của sản phẩm phù hợp với tiêu chuẩn đã được duyệt; theo dõi hiệu năng của các chất chuẩn, chất tham chiếu, của các thử nghiệm. |
- 1Circular No. 11/2018/TT-BYT dated May 4, 2018
- 2Integrated document No. 06/VBHN-BYT dated July 03, 2020 Circular on quality of pharmaceutical products and pharmaceutical starting materials
- 3Integrated document No. 06/VBHN-BYT dated July 03, 2020 Circular on quality of pharmaceutical products and pharmaceutical starting materials
- 1Circular No. 9/2020/TT-BYT dated June 10, 2020 on amendments to the Circular No. 03/2018/TT-BYT on good distribution practices for pharmaceutical products and pharmaceutical starting materials
- 2Decree No. 155/2018/ND-CP dated November 12, 2018 on amendments related to business conditions under state management of the Ministry of Health
- 3Circular No. 32/2018/TT-BYT dated November 12, 2018
- 4Decree No. 75/2017/ND-CP dated June 20, 2017, defining functions, tasks, powers and organizational structure of Ministry of Health
- 5Decree No. 54/2017/ND-CP dated May 08, 2017, guidelines for implementation of the Law on Pharmacy
- 6Law No. 105/2016/QH13 dated April 06th, 2016, on pharmacy
- 7Joint Circular No. 58/2015/TTLT-BYT-BTNMT dated December 31st, 2015, stipulating regulations on biomedical waste management
- 8Circular No. 36/2015/TT-BTNMT dated June 30, 2015, management of hazardous wastes
- 9Law No. 01/2011/QH13 of November 11, 2011 on Archives
Circular No. 03/2020/TT-BYT dated January 22, 2020 on amendments to Circular No. 11/2018/TT-BYT on quality of pharmaceutical products and pharmaceutical starting materials
- Số hiệu: 03/2020/TT-BYT
- Loại văn bản: Thông tư
- Ngày ban hành: 22/01/2020
- Nơi ban hành: Bộ Y tế
- Người ký: Trương Quốc Cường
- Ngày công báo: Đang cập nhật
- Số công báo: Đang cập nhật
- Ngày hiệu lực: 16/03/2020
- Tình trạng hiệu lực: Kiểm tra
Đơn vị chủ quản: Công ty cổ phần tư vấn đầu tư và ứng dụng công nghệ 4.0.
Chịu trách nhiệm chính: Bà Phạm Hoài Thương.
Giấy chứng nhận ĐKDN số: 0108234370, do Sở Kế hoạch và Đầu tư thành phố Hà Nội cấp ngày 18/04/2018.
Địa chỉ: Thôn Trung, Xã Phù Đổng, TP Hà Nội - VPGD: C2 Vincom, 119 Trần Duy Hưng, Phường Yên Hòa, TP Hà Nội.
Điện thoại: 024.6294.9155 - Hotline 1: 0342.799.688 - Hotline 2: 0985.426.175 - Email: info@hethongphapluat.com

